Signed in as:
filler@godaddy.com

As outside collaborator, we provide computational chemistry support for the the research program entitled "Probing novel pathways of metal sulfide acquisition and trafficking from minerals in model biocatalytic systems" hby the DOE EPSCoR program as the Phase II proposal for the DOE-EPSCoR team directed by Prof. Eric Boyd.
We explore the chemical space illustrated above that connects the abiotic pyrite reduction and dissolution and the biotic Fe-S cluster acquisition in methanogens. Please contact us if you are interested in learning about Fe-S geochemistry, physical chemistry, and biochemistry..

As part of the international consortia "Atomic Design of Carbon Materials" (AtomDeC) and in collaboration with Prof. Nicholas Stadie, we are exploring the emerging properties of porous carbon materials with heteroatom doping for gas adsorption, electronic devices using computational modeling tools that range from molecular mechanics to ab initio quantum dynamics methods.
Our team contributes to Work Packet 4 by considering molecular cluster modells, maquettes of carbon materials with ill-defined structure and composition. Please visit the AtomDeC website for more information about the team composition, events, and open positions.

A long time interest of the lab is the computational and experimental evaluations of various chemical scenarios for the emergence of the building blocks of biochemistry. In addition to Fe-S chemistry, we are also focused on condensation and hydrolysis reactions that could give rise to the formation of macromolecules as precursors for biopolymers.

The above image from Encyclopaedia Britannica illustrates the N-cycle on Earth. Careful evaluation of all the redox pathways for interconversion of N species reveals a missing reaction, which is ammonia oxidation to N2. This reaction could take place under anoxic conditions in the presence of radical species that initiate the process by H-atom abstraction.
Given the high-energy content of N-H bonds, ammonia molecules or ammonium ions are potential ecology friendly fuels for driving both human technology and extant biology. It is a fascinating idea to consider whether biochemical pathways utilizing ammonia as energy source went extinct during the geological eons of Earth evolution. In collaboration with Prof. Michael Mock we are exploring the potential energy surface of ammonia interconversion to nitrogen gas.
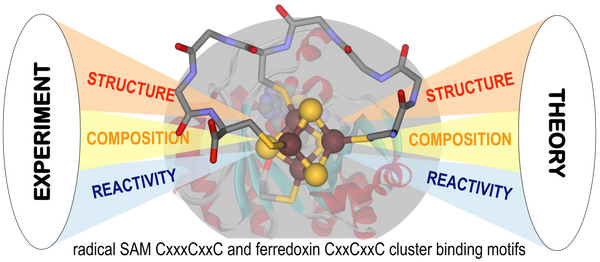
Synthetic Biomimetic Design of Radical SAM Maquettes from Experiment and Theory
The research aims to construct maquettes of SAM radical enzymes with a minimalist CxxxCxxC binding motif to house a redox active [4Fe-4S] cluster and coordinate S-adenosyl-methionine cofactor as Nature’s most versatile bioinorganic system for radical-based biochemical transformations in a combined synthetic, spectroscopic, and computational research approach.

Clay Embedded Fe-S Clusters: Synthesis, Characterization, and Catalytic Application
A synthetic platform is developed for revealing fundamental compositional and structural guidelines in order to define catalytic function in site differentiated nanokaolinite molecules. The synthetic and reactivity studies are complemented with comprehensive spectroscopic characterization and computational chemistry using realistic molecular models.
Quantum Chemical Engineering of Kaolinite Nanoparticles and their Reactivity
Computational methodologies are established for exfoliated kaolinite nanoparticles in a “bottom-up” approach using empirical force field and semi- empirical quantum chemical methods. These methods together with ab initio density functional theory calculations enable to construct integrated, multi-layered computational models for atomic level description of clay reactivity.
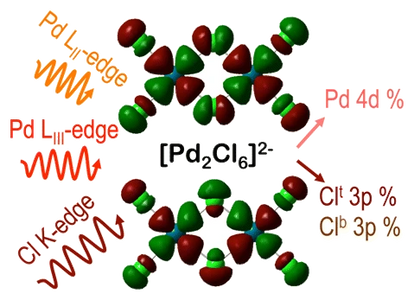
Multi-edge X-ray Absorption Spectroscopy for Catalyst Characterization and Design
A “turn-key” synchrotron-based spectroscopic technique designed for organic and organometallic chemists can provide new insights into transition metal-based transformations. The spectroscopic data validate ab initio density functional theory with respect to ground state, structural information with the goal of defining atomic-level details of molecular mechanisms.